ChIP-Seq 数据分析流程简介
真核生物的在基因表达时,需要大量的转录因子辅助。真和生物的很多生理反应都是通过转录因子。染色质免疫共沉淀技术(Chromatin Immunoprecipitation,ChIP)也称结合位点分析法,是研究体内蛋白质与DNA相互作用的有力工具,通常用于转录因子结合位点或组蛋白特异性修饰位点的研究。
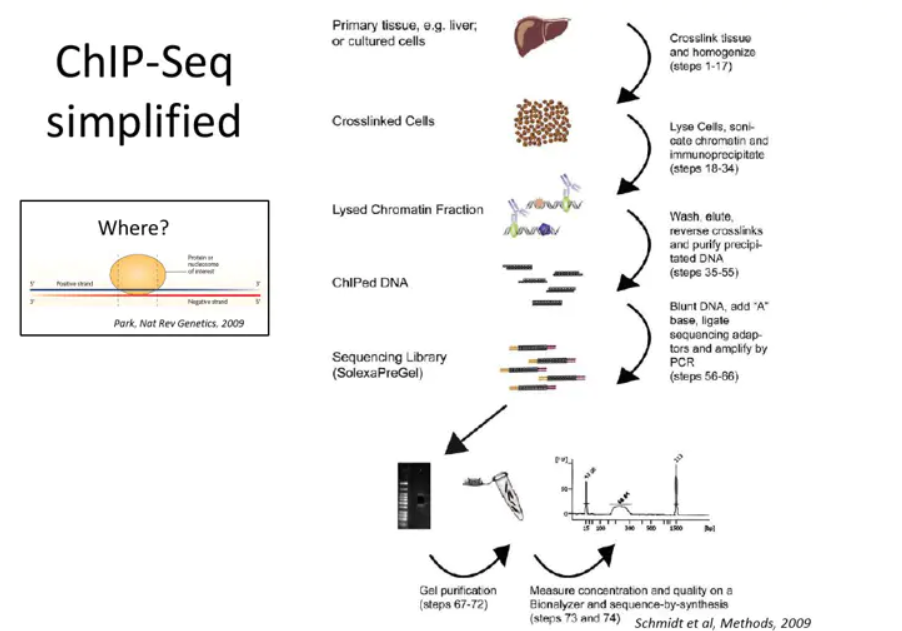
1 质量控制
首先采用Trimmomatics 工具对低质量和存在接头的序列进行清洗,去除后的clean数据可以进一步分析。数据质量可以先用fastqc快速可视化。
2 序列比对
采用bowtie2(很多文章他会提到read uniquely mapped 来进行下游分析,bowtie可以通过设置-m 1 参数,但是对于bowtie2以及bwa,并不存在这样的参数,可以使用bwa输出中的XT:A:U(用于标记unique hit)以及tophat中的NH:i:1(用于标记unique hit)来进行过滤,bowtie2则可以根据sam或bam文件的tag来提取)
3 peak calling
MACS分析,用于鉴定转录因子结合位点。 MACS抓住了基因组复杂性对评估其重要性的影响丰富的ChIP区域,MACS提高了空间分辨率通过组合两个测序标签的信息来结合位点位置和方向。 MACS可以很容易地用于ChIP-Seq数据单独使用,或与对照样品一起增加特异性。
4 Motif注释
转录因子motif是一些很短的模序(~10bp),在大基因组里很容易出现随机比对,而且是以position weight matrix (PWM)格式来呈现,说明它的可变性,因此研究motif有哪些binding区域是没有意义的,因为很难找到一个方法(阈值)来判断真正的比对和随机的比对。
HOMER采用的思路是:给定一个指定的区域(promoter或enhancer区域),根据统计学检验,我们很容易知道一个motif是否显著富集在这个区域(与背景区域相比)。