scATAC分析
编者按:转录调控是基础科研领域非常重要的研究方向,深入浅出的介绍了单细胞测序技术在ATAC-Seq 方向上的应用。在此感谢生信技能树的单细胞天地的本文作者 Kinesin。
1. scATAC简介
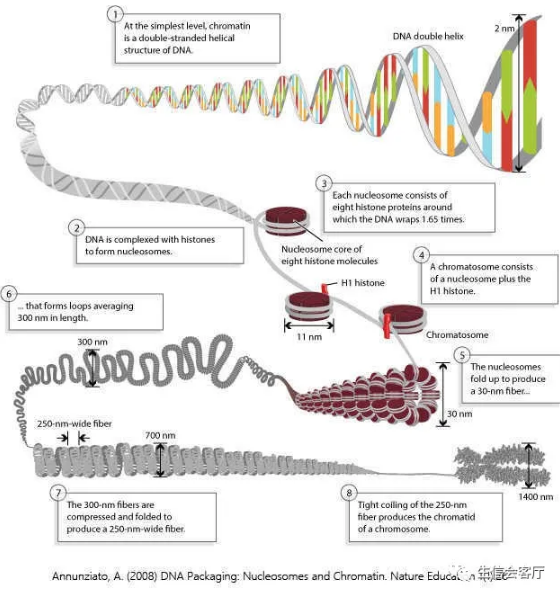
高级生物中DNA以层层折叠的形式压缩在染色体中,这种状态下的DNA通常不能复制、转录或结合其他调控因子。我们知道细胞内时刻都有转录调控作用发生,那是怎么实现的呢?细胞会通过组蛋白修饰“解压”部分染色质(通常占总体的2-3%)让DNA暴露出来,这样各种转录调控因子就可以结合上来发挥作用了,这种开放的DNA区域也叫做染色质可及性(chromatin accessibility)区域。
2013年美国斯坦福大学的William Greenleaf教授研发了一种全新的方法,利用DNA转座酶结合高通量测序技术来研究染色体的可及性,即ATAC-seq (Assay for transposase-accessible chromatin with high-throughput sequencing)。
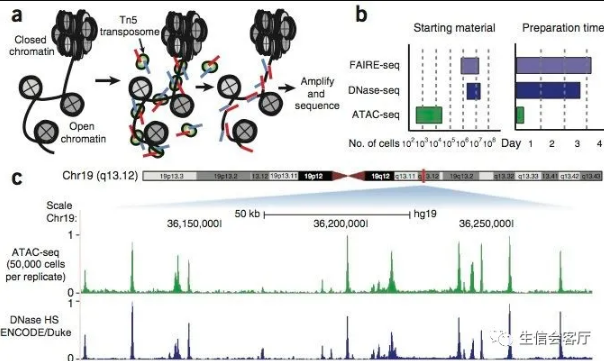
DNA转座,是一种把DNA序列从染色体的一个区域搬运到另外一个区域的现象,由DNA转座酶来实现。这种转座插入DNA,也是需要插入位点的染色质是开放的,否则就会被一大坨高级结构给卡住。那么,如上图a,我们只要人为地,将携带已知DNA序列标签的转座复合物(即带着红色蓝色测序标签的转座酶Tn5),加入到细胞核中,再利用已知序列的标签进行PCR后测序,就知道哪些区域是开放染色质了。而这也就是ATAC-seq的原理。
2. 10X scATAC-seq的简介
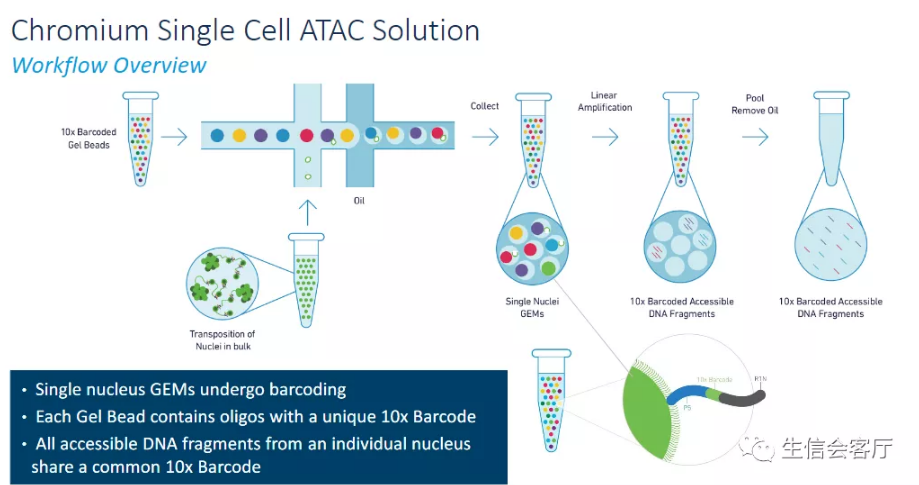
10X Genomics公司的scATAC-seq是目前主流的单细胞水平染色质可及性测序解决方案,国内已经有很多公司推出了商业化服务。该技术基于10X Genomics Chromium平台,可用于绘制细胞染色质开放区的单细胞图谱,是一种单细胞水平研究表观遗传学的有效手段。其工作流程如下:
- 使用改造过的Tn5转座酶孵育细胞核,Tn5转座酶剪切开放区域DNA并加上测序接头;
- 利用微流控系统将带有barcode引物的凝胶微珠和单个细胞核包裹在油滴中;
- 油滴内凝胶微珠溶解释放出带barcode的引物,细胞核裂解释放带有测序接头的染色质开放区域DNA,引物与DNA结合进行PCR扩增;
- 油层破裂,DNA片段释放出来,进行后续的Illumina文库构建和测序。
由于每个凝胶微珠的barcode序列具有唯一性,凝胶微珠与细胞核也是唯一配对关系,所以可以通过barcode识别哪些DNA序列来自同一个细胞核,我们也就得到了单细胞水平的ATAC-seq数据。
3. scATAC-seq的意义
染色质开放区域含有多种调节元件:启动子,增强子,绝缘子等。ATAC-seq和scATAC-seq得到的数据中,染色质开放区域含有更多的reads。通过生物信息学分析峰与转录起始位点的距离,可以得到处于开放区域作用下的基因列表;或分析开放区域内的motif,得到活跃基因的上游调控因子,提供全基因组转录因子发生的信息。scATAC-seq相比ATAC-seq的独特优势:
- 在单细胞水平揭示细胞核内染色质可及性;
- 揭示染色质可及性在细胞间的异质性;
- 鉴定特定细胞类型的染色质活性区域;
4. scRNA与scATAC的整合分析
DNA层面的调控与mRNA层面的表达存在因果关系,因此整合两种类型的数据既可以相互印证研究结论,也更容易找到完整的调控网络环路。此外,在疾病研究中使用多组学数据基于主效应或交互作用筛选生物标志物可提高疾病预后的准确性。
4.0 下载分析数据
这次分析使用10x 官方提供的pbmc示例数据,既有scRNA数据又有scATAC数据,下载链接如下:
| 数据 | 下载地址 |
|---|---|
| scATAC-seq peak矩阵 | http://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5 |
| scATAC-seq metadata | http://cf.10xgenomics.com/samples/cell-atac/1.0.1/atac_v1_pbmc_10k/atac_v1_pbmc_10k_singlecell.csv |
| scRNA-seq seurat对象 | https://www.dropbox.com/s/3f3p5nxrn5b3y4y/pbmc_10k_v3.rds?dl=1 |
| GRCh37基因组注释文件 | ftp://ftp.ensembl.org/pub/grch37/release-84/gtf/homo_sapiens/Homo_sapiens.GRCh37.82.gtf.gz |
scRNA-seq seurat对象文件是降维聚类并鉴定细胞类型的scRNA数据,下载困难的朋友可以下载官方的表达矩阵自己处理。 scRNA-seq 表达矩阵 http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k_v3_filtered_feature_bc_matrix.h5
4.1 scATAC数据预处理
library(Seurat)
library(ggplot2)
library(rtracklayer)
library(patchwork)
rm(list=ls())
dir.create("ATAC")
set.seed(811)
peaks <- Read10X_h5("Data/ATAC/atac_v1_pbmc_10k_filtered_peak_bc_matrix.h5")
scATAC-seq得到的初始文件是peak矩阵,行为染色体坐标,列为细胞barcode,这些染色体坐标代表染色质可及性区域。
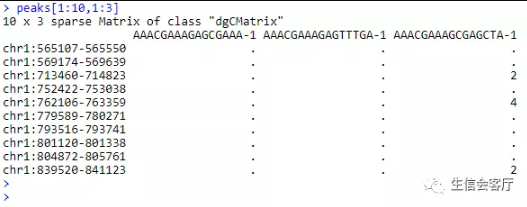
我们假设基因上游0~2kb区域的reads数可以代表基因的活性,并把上面的peak矩阵转换为基因活性矩阵。
activity.matrix <- CreateGeneActivityMatrix(peak.matrix = peaks,
annotation.file = "Data/ATAC/Homo_sapiens.GRCh37.82.gtf",
seq.levels = c(1:22, "X", "Y"), upstream = 2000, verbose = TRUE)
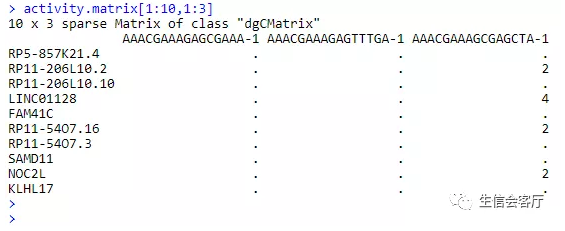
使用peak矩阵、活性矩阵和metadata创建seurat对象
pbmc.atac <- CreateSeuratObject(counts = peaks, assay = "ATAC", project = "10x_ATAC")
pbmc.atac[["ACTIVITY"]] <- CreateAssayObject(counts = activity.matrix)
meta <- read.table("Data/ATAC/atac_v1_pbmc_10k_singlecell.csv", sep = ",",
header = TRUE, row.names = 1, stringsAsFactors = FALSE)
meta <- meta[colnames(pbmc.atac), ]
pbmc.atac <- AddMetaData(pbmc.atac, metadata = meta)
pbmc.atac <- subset(pbmc.atac, subset = nCount_ATAC > 5000)
pbmc.atac$tech <- "atac"
使用peak矩阵对scATAC数据降维,并对活性矩阵进行标准化
#对活性矩阵执行标准化与中心化处理
DefaultAssay(pbmc.atac) <- "ACTIVITY"
pbmc.atac <- FindVariableFeatures(pbmc.atac)
pbmc.atac <- NormalizeData(pbmc.atac)
pbmc.atac <- ScaleData(pbmc.atac)
#对peak矩阵降维
DefaultAssay(pbmc.atac) <- "ATAC"
VariableFeatures(pbmc.atac) <- names(which(Matrix::rowSums(pbmc.atac) > 100))
pbmc.atac <- RunLSI(pbmc.atac, n = 50, scale.max = NULL)
pbmc.atac <- RunUMAP(pbmc.atac, reduction = "lsi", dims = 1:50)
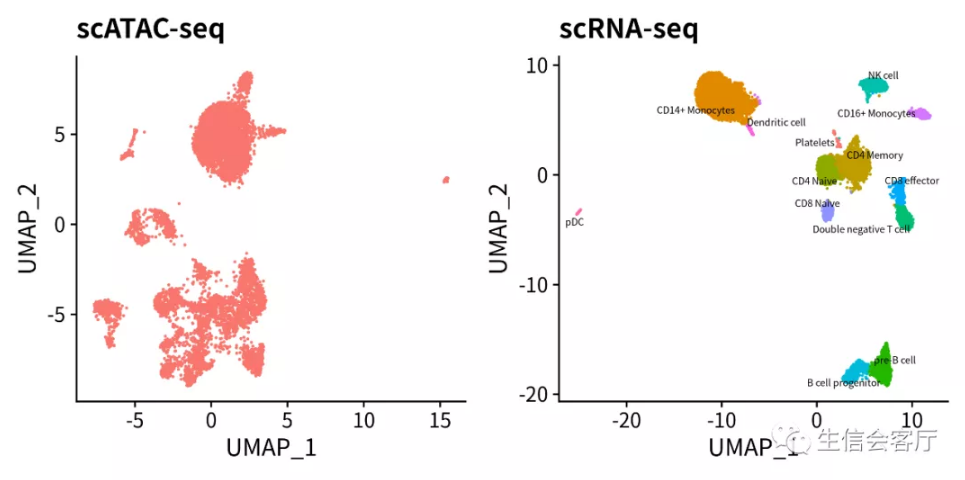
4.2 整合前scRNA与scATAC数据展示
pbmc.rna <- readRDS("Data/ATAC/pbmc_10k_v3.rds")
pbmc.rna$tech <- "rna"
#展示整合前的scATAC和scRNA数据
p1 <- DimPlot(pbmc.atac, reduction = "umap") + NoLegend() + ggtitle("scATAC-seq")
p2 <- DimPlot(pbmc.rna, group.by="celltype", label=TRUE, label.size=2,
repel = TRUE) + NoLegend() + ggtitle("scRNA-seq")
plotc = p1 + p2
ggsave('ATAC/ATAC-RNA_beforeINT.png', plotc, width = 8, height = 4)
4.3 识别scATAC数据集的细胞类型
在单细胞转录组高级分析一:多样本合并与批次校正中,我们介绍过Seurat可以将两个数据集联系起来,并通过“锚点”找到两个数据集之间的同类细胞。找到同类细胞之后不仅可以使用“差异向量”校正批次效应,还可以实现两个数据集之间的数据转换。Seurat将需要进行数据转换的两个数据集,一个称为查询数据集,一个称为参考数据集。参考数据集和查询数据集之间建立细胞匹配关系后,可以将参考数据集细胞的类型、元数据、转录谱等信息迁移给配对的查询数据集细胞。
用已注释细胞类型的scRNA数据集作为参考数据集,通过上述信息迁移的方法就可以识别scATAC数据集的细胞类型。
#通过锚点建立数据集之间的配对关系
transfer.anchors <- FindTransferAnchors(reference=pbmc.rna, query = pbmc.atac,
features=VariableFeatures(object=pbmc.rna), reference.assay = "RNA",
query.assay = "ACTIVITY", reduction = "cca")
#数据迁移
celltype.predictions <- TransferData(anchorset = transfer.anchors, refdata = pbmc.rna$celltype,
weight.reduction = pbmc.atac[["lsi"]])
pbmc.atac <- AddMetaData(pbmc.atac, metadata = celltype.predictions)
#展示id转换后的scATAC和scRNA数据
pbmc.atac.filtered <- subset(pbmc.atac, subset = prediction.score.max > 0.5)
pbmc.atac.filtered$predicted.id <- factor(pbmc.atac.filtered$predicted.id, levels=levels(pbmc.rna))
p1 <- DimPlot(pbmc.atac.filtered, group.by="predicted.id", label=TRUE, label.size=2, repel=TRUE) +
ggtitle("scATAC-seq cells") + NoLegend() + scale_colour_hue(drop = FALSE)
p2 <- DimPlot(pbmc.rna, group.by="celltype", label=TRUE, label.size=2, repel=TRUE) +
ggtitle("scRNA-seq cells") + NoLegend()
plotc = p1 + p2
ggsave('ATAC/ATAC-RNA_transfer.png', plotc, width = 8, height = 4)
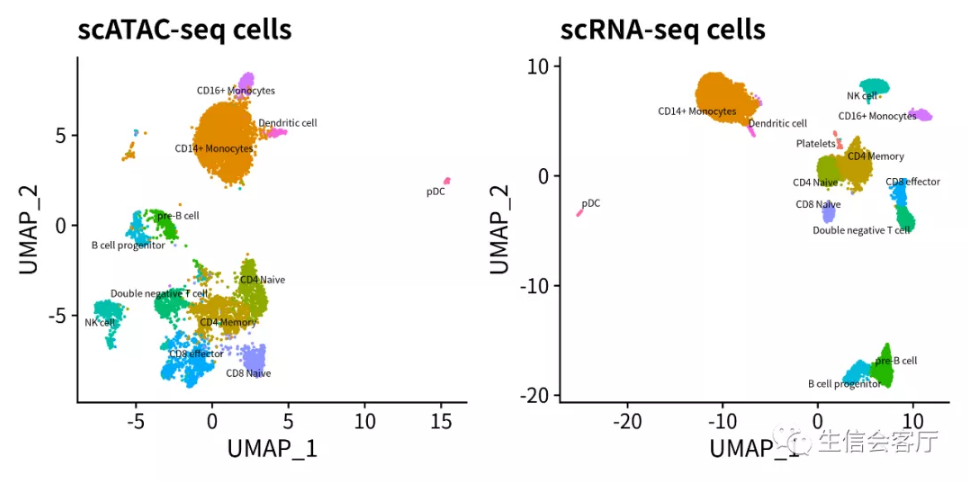
经过上面的细胞类型识别,我们就能同时得到某类细胞的RNA数据和ATAC数据,进而开展多组学联合分析。
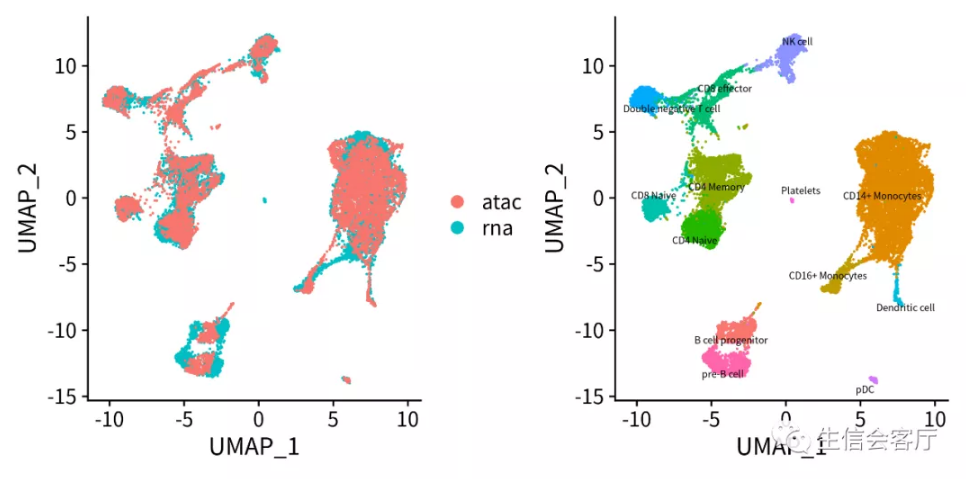
4.4 观察两个数据集的异同
将scRNA数据集中细胞的转录谱迁移给scATAC数据集的配对细胞,然后对合并的数据集进行降维,就可以实现两个数据集中的配对细胞的共定位。
#将scRNA细胞的转录谱通过锚点转移给scATAC细胞
genes.use <- VariableFeatures(pbmc.rna)
refdata <- GetAssayData(pbmc.rna, assay="RNA", slot="data")[genes.use, ]
imputation <- TransferData(anchorset=transfer.anchors, refdata=refdata, weight.reduction = pbmc.atac[["lsi"]])
pbmc.atac[["RNA"]] <- imputation
#合并两个数据集的seurat对象
coembed <- merge(x = pbmc.rna, y = pbmc.atac)
#合并后的数据集执行降维分析
coembed <- ScaleData(coembed, features = genes.use, do.scale = FALSE)
coembed <- RunPCA(coembed, features = genes.use, verbose = FALSE)
coembed <- RunUMAP(coembed, dims = 1:30)
coembed$celltype <- ifelse(!is.na(coembed$celltype), coembed$celltype, coembed$predicted.id)
p1 <- DimPlot(coembed, group.by = "tech")
p2 <- DimPlot(coembed, group.by="celltype", label=TRUE, label.size=2, repel=TRUE) + NoLegend()
plotc = p1 + p2
ggsave('ATAC/ATAC-RNA_coembed.png', plotc, width = 8, height = 4)
观察上图我们可以发现,绝大部分细胞类型两者都会重叠,但是也有各自独有的cluster。例如血小板只出现在scRNA-seq数据中,这是因为它们是骨髓内成熟巨核细胞胞质脱离产生,所以scATAC检测不到相关数据。因此,不要期望scRNA与scATAC在分析中完全一致。