基因组组装基础套路
以Pacbio和Nanopore 为代表的三代测序技术,目前最成功的应用领域就是基因组组装,由于其长读长的特点,非常适合处理对于二代测序技术较难测通的高重复区域和高GC含量区域。因此对于基因组组装研究而言,三代测序技术已经成为了现实意义上的标准技术。但是相伴产生的问题就是计算量的大幅增长。笔者早年在测序公司工作期间,第一个任务就是搭建宏基因组组装流程,期间由于内存超标造成的不断失败的依然历历在目。
本文的主要目的,是从一个基础的基于三代测序数据(Pacbio为例)为基础的组装策略,展示一下三代基因组组装的一些难关。
第一关 k-mer 分析
基因组组装第一步就是需要对需要组装的物种基因组大小、测序样本杂合度信息有一个估计,一般采用k-mer 分析作为基础。k-mer分析简单来说,就是将所测数据中长度为k的碱基序列频率进行统计,从k-mer出现频率分布情况,估算样本的杂合程度、基因组大小等信息。该步骤一般采用JellyFish 软件进行操作。由于该步骤是一个对内存消耗较大的。
而且,由于k-mer分析中的k值其实没有一个确定值,因此在实际分析过程中,经常会对
第二关 长序列数据矫正
三代测序在提供了长读长的基础上也损失了一定的准确性(与二代测序相比),而二代测序相对低廉的价格以及较高的准确性,也非常适合与三代测序数据结合,进行reads的矫正。除此之外,在常见的三代组装软件之中(例如FALCON、CANU等)其内部其实存在一个三代序列自我矫正的步骤,但从笔者有限的经验来看,如果不在组装之前进行矫正,后面的组装结果依然是惨不忍睹。另外从资金成本角度上来看,一味地增加三代数据量和增加二代测序数据相比也是不划算的。因此采用二代数据对三代数据进行矫正就显得十分合理了(当然,土豪请忽略…)
三代序列自矫正工具LoRMA,
ARAMIS是二代测序数据纠正三代数据的工具。
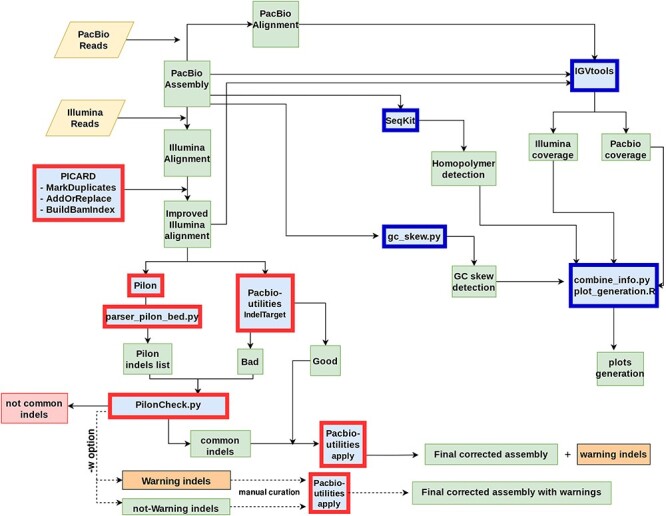
该步骤的核心其实是序列比对运算,因此该步骤也是对内存消耗十分巨大的步骤之一。
第三关 基因组组装计算
三代基因组组装常用的工具有FALCON和canu这类工具基本上就一个特点:占用大量内存。以FALCON为例,该工具已经成为Pacbio 官方发布的分析框架SMRT link的一个标准工具,其分析的每一步,都伴随着将大量数据读入内存进行后续计算。而如果样本本身的杂合性较高,内存需求也会较杂合度较小的数据相比有较大的提升。因此,对于组装计算这步而言,内存的巨大消耗是目前无法绕过去的坎。
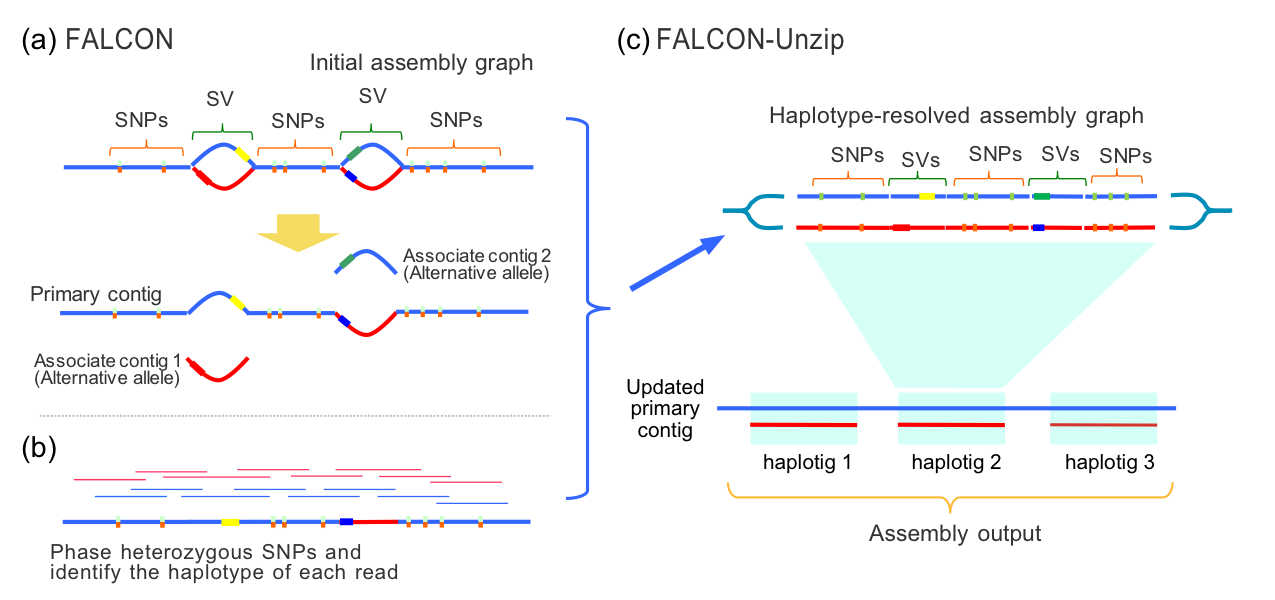
当然,二代测序数据组装与三代组装相比只能是更加占用内存,这主要是因为二代组装软件(如华大基因开发的SOAPdenovo),基本上都是基于德布鲁因图(de Bruijn graph)算法的,该算法对内存占用,比三代主流的以重叠区域为核心的算法,对占用情况更大。
不仅如此,二代测序中最为常见的Call SNP操作的最知名的工具集——GATK,在其算法核心中,其实也有通过局部组装来矫正比对结果的步骤,因此在不能并发的条件下,单个线程对所对应的内存也是非常感人的…
总之,在目前的测序技术的条件下,组装(除了基因组之外,也包括无参转录组),在一般的生信分析中都是绝对的内存杀手。
小结
三代基因组组装是涉及的所有步骤,基本上都是十分占用内存的任务。类似的,二代数据无参转录组组装也是一类常见的组装问题,对于内存同样有较高的要求。这就造成了对于多节点的计算环境而言,一般需要配置胖节点进行组装运算。但是胖节点不能总为组装任务保持空闲着,所以有效的任务调度又成为了组装任务的潜在关卡。
在生物信息相关计算中,由于计算的特殊性造成了对单个任务占用大量内存和有限的硬件资源之间存在天然的矛盾,因此解决之道,在于采用更合理的作业调度和资源调度系统。