比较基因组学3: Spark 计算框架加速多重序列比对 —— HAlign
多重序列比对(Multiple Sequences Alignment MSA )是进化分析最为基础的操作之一,其主要功能是揭示不同物种或个体之间的序列差异。同时,多重序列比对也是进化树重构的重要步骤之一。
但是多重序列比对也是公认的生物信息学计算瓶颈之一。由于计算量大,因此在处理大型数据集(序列数量多)时,常常存在困难。目前已经有不少从计算框架角度解决这一问题的尝试,例如:ClustalW-MPI。本次向各位读者介绍的,是基于Spark框架运行HAlign 程序进行多重序列比对的方式。
HAlign 原理
HAlign 是我国天津大学团队开发的一款针对DNA/RNA序列的多重序列比对工具,更多信息可以通过其官网获取。HAlign 通过前缀树算法实现了多重序列比对问题进行拆分,使得问题可以通过Mapreduce 方式进行并行加速。其基本原理如下图所示:
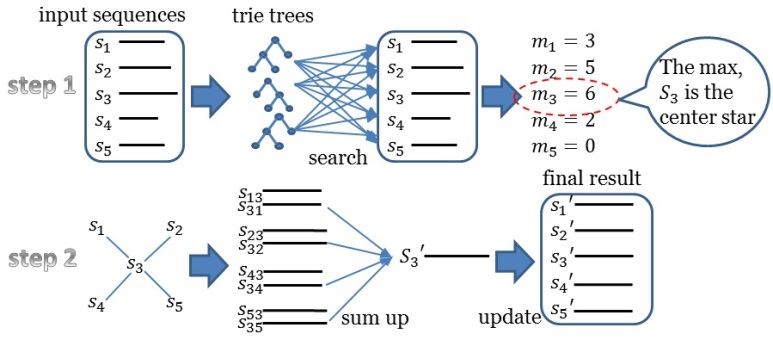
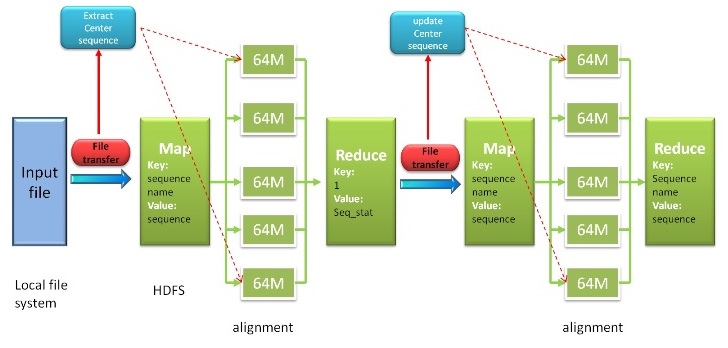
在Achelous 平台上运行 HAlign 进行多重序列比对计算
用户可以通过Achelous的 WDL 语法中,对Spark 框架的支持,实现分布式运行HAlign任务。示例wdl 脚本如下:
workflow msa_spark{
File input_fasta
String prefix
String SparkExecutorURI
call halign_spark {
input:
input_fasta = input_fasta,
prefix = prefix,
SparkExecutorURI = SparkExecutorURI
}
}
task halign_spark {
File input_fasta
String prefix
String SparkExecutorURI = "hdfs://Cc8Apc:9000/xtao-internal/spark-2.2.0-bin-hadoop2.7.tgz"
command <<<
spark-submit --master ${SPARK_MASTER_URI} --num-executors 8 --executor-cores 2 --executor-memory 90g --driver-memory 30g --conf 'spark.cores.max=16' --class main /bio/HAlign-2.1.jar -sparkMSA ~{input_fasta} ~{prefix}.aligned.fa 0
>>>
output {
File aligned_fasta = "${prefix}.aligned.fa"
}
runtime {
docker: "spark-halign:2.1"
cpu: "2"
memory: "20G"
SparkExecutorURI: "${SparkExecutorURI}"
usextaosparkscheduler: "true"
ldapauth: true
volumes: {
"/var/spark/storage" : "/var/spark/storage"
}
}
}
用户添加对应流程后,即可使用该流程进行多重序列比对分析。
在Achelous 上使用 Spark框架进行编程
与MPI分布式框架相比,Spark 框架具有较好的开发接口,对开发者的编程能力要求也相对友好。对于 Achelous 平台用户而言,可以在系统部署后,轻松实现多种分布式计算框架的使用。